Examining and creating files
Approximate time: 30 min
Learning Objectives
- View the contents of a file
- Create a new file using the Nano text editor
- Execute basic shortcuts in the Nano text editor
Examining Files
We can navigate directories — now let's look at the contents of files using a few key commands.
cat command
The easiest way to examine a file is to just print out all of its contents using the command cat. We can test this out by printing the contents of ~/unix_lesson/other/sequences.fa. Enter the command followed by the filename, including the path when necessary:
$ cat ~/unix_lesson/other/sequences.fa
The cat command prints all contents of the file to the screen.
What does this file contain?
>SRR014849.1 EIXKN4201CFU84 length=93
GGGGGGGGGGGGGGGGCTTTTTTTGTTTGGAACCGAAAGGGTTTTGAATTTCAAACCCTTTTCGGTTTCCAACCTTCCAAAGCAATGCCAATA
>gi|340780744|ref|NC_015850.1| Acidithiobacillus caldus SM-1 chromosome, complete genome
ATGAGTAGTCATTCAGCGCCGACAGCGTTGCAAGATGGAGCCGCGCTGTGGTCCGCCCTATGCGTCCAACTGGAGCTCGTCACGAG
TCCGCAGCAGTTCAATACCTGGCTGCGGCCCCTGCGTGGCGAATTGCAGGGTCATGAGCTGCGCCTGCTCGCCCCCAATCCCTTCG
TCCGCGACTGGGTGCGTGAACGCATGGCCGAACTCGTCAAGGAACAGCTGCAGCGGATCGCTCCGGGTTTTGAGCTGGTCTTCGCT
CTGGACGAAGAGGCAGCAGCGGCGACATCGGCACCGACCGCGAGCATTGCGCCCGAGCGCAGCAGCGCACCCGGTGGTCACCGCCT
CAACCCAGCCTTCAACTTCCAGTCCTACGTCGAAGGGAAGTCCAATCAGCTCGCCCTGGCGGCAGCCCGCCAGGTTGCCCAGCATC
CAGGCAAATCCTACAACCCACTGTACATTTATGGTGGTGTGGGCCTCGGCAAGACGCACCTCATGCAGGCCGTGGGCAACGATATC
CTGCAGCGGCAACCCGAGGCCAAGGTGCTCTATATCAGCTCCGAAGGCTTCATCATGGATATGGTGCGCTCGCTGCAACACAATAC
CATCAACGACTTCAAACAGCGTTATCGCAAGCTGGACGCCCTGCTCATCGACGACATCCAGTTCTTTGCGGGCAAGGACCGCACCC
>gi|129295|sp|P01013|OVAX_CHICK GENE X PROTEIN (OVALBUMIN-RELATED)
QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAE
less command
For large files, cat floods the terminal. Use less instead to page through the file.
Move into our raw_fastq directory and enter the following command:
$ less Mov10_oe_1.subset.fq
less opens the file in a scrollable buffer. Use these shortcuts:
Shortcuts for less
| key | action |
|---|---|
| SPACE | to go forward |
| b | to go backwards |
| g | to go to the beginning |
| G | to go to the end |
| q | to quit |
Use the shortcut keys to move through your FASTQ file, we will explore these files in more detail later in the workshop.
Searching files with less
Type / followed by a search term to find text in the file. Press /+ENTER to repeat the last search. Hit q to exit.
head and tail commands
There’s another way that we can look at files, and just look at part of them. In particular, if we just want to see the beginning or end of the file to see how it’s formatted.
The commands are head and tail and they just let you look at the beginning and end of a file respectively.
$ head Mov10_oe_1.subset.fq
$ tail Mov10_oe_1.subset.fq
By default, the first or last 10 lines will be printed to screen. The -n option can be used with either of these commands to specify the number n lines of a file to display. For example, let’s print the first/last line of the file:
$ head -n 1 Mov10_oe_1.subset.fq
$ tail -n 1 Mov10_oe_1.subset.fq
Exercise
- Change directories into
genomics_data. You can do this using a full or relative path. - Use the
lesscommand to open up the fileEncode-hesc-Nanog.bed. - Search for the string
chr11; you’ll see all instances in the file highlighted. - Staying in the
lessbuffer, use the shortcut to get to the end of the file. Report the three highlighted lines at the end of the file where you seechr11highlighted. - Exit the
lessbuffer and come back to the command prompt. - Print to screen the last 5 lines of the file
Encode-hesc-Nanog.bed. Report what you see as the output within the Terminal.
Writing files
To create or edit files on the command line we need a text editor. We’ll use nano — a simple, keyboard-driven editor that works on any remote system.
Introduction to Nano
To write and edit files, we’re going to use a text editor called ‘Nano’. Nano is a simple, yet powerful tool for editing text files directly from the command line.
1. Introduction to Nano
Nano is a user-friendly text editor designed for Unix-like systems, such as Linux. It is often pre-installed on many distributions and is known for its simplicity and ease of use. Unlike more complex editors like Vim or Emacs, Nano provides a straightforward interface with on-screen shortcuts, making it an excellent choice for beginners.
In this course, you'll learn how to use Nano to create and edit files, navigate its interface, and perform common tasks efficiently.
2. Opening Nano
To start using Nano, open your terminal and type:
nanoThis opens Nano with a new, empty file. To edit an existing file or create a new one with a specific name, type:
nano filename.txtIf filename.txt doesn't exist, Nano will create it when you save your work.
3. Basic Navigation
When you open Nano, you'll see a text area where you can type or edit content. At the bottom, there are two lines of shortcuts, where ^ represents the Ctrl key (e.g., ^G means Ctrl + G). Here's what the Nano interface looks like:
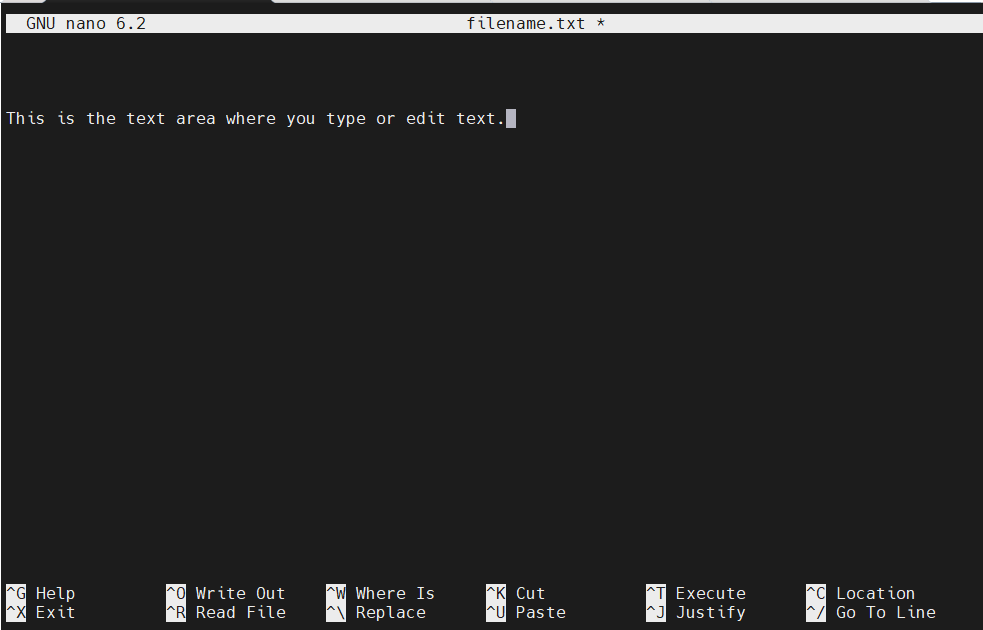
Use the arrow keys to move the cursor around the text. Additional navigation shortcuts include:
Ctrl + A: Move to the start of the current line.Ctrl + E: Move to the end of the current line.Ctrl + Y: Scroll up one page.Ctrl + V: Scroll down one page.
4. Editing Text
To add text, simply start typing. To remove text, use:
- Backspace: Delete characters before the cursor.
- Delete: Delete characters after the cursor.
For cutting and pasting:
Ctrl + K: Cut the entire line where the cursor is.Ctrl + U: Paste the cut text at the cursor's position.
5. Saving and Exiting
To save your changes, press Ctrl + O. Nano will prompt you to confirm the filename at the bottom of the screen, like this:
Press Enter to save. To exit Nano, press Ctrl + X. If you have unsaved changes, Nano will ask if you want to save them before closing.
This lesson has been modified from a course developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.